The evolutionary algorithm
A Bradley Duthie
2026-05-28
Source:vignettes/evolutionary_algorithm.Rmd
evolutionary_algorithm.RmdIntroduction
The resevol package models individuals with complex genomes that can include any number of haploid or diploid loci and any number of traits with an arbitrary pre-specified covariance structure. It does this by using a complex network mapping a path from the allele values at each loci to the covarying trait values of individuals.
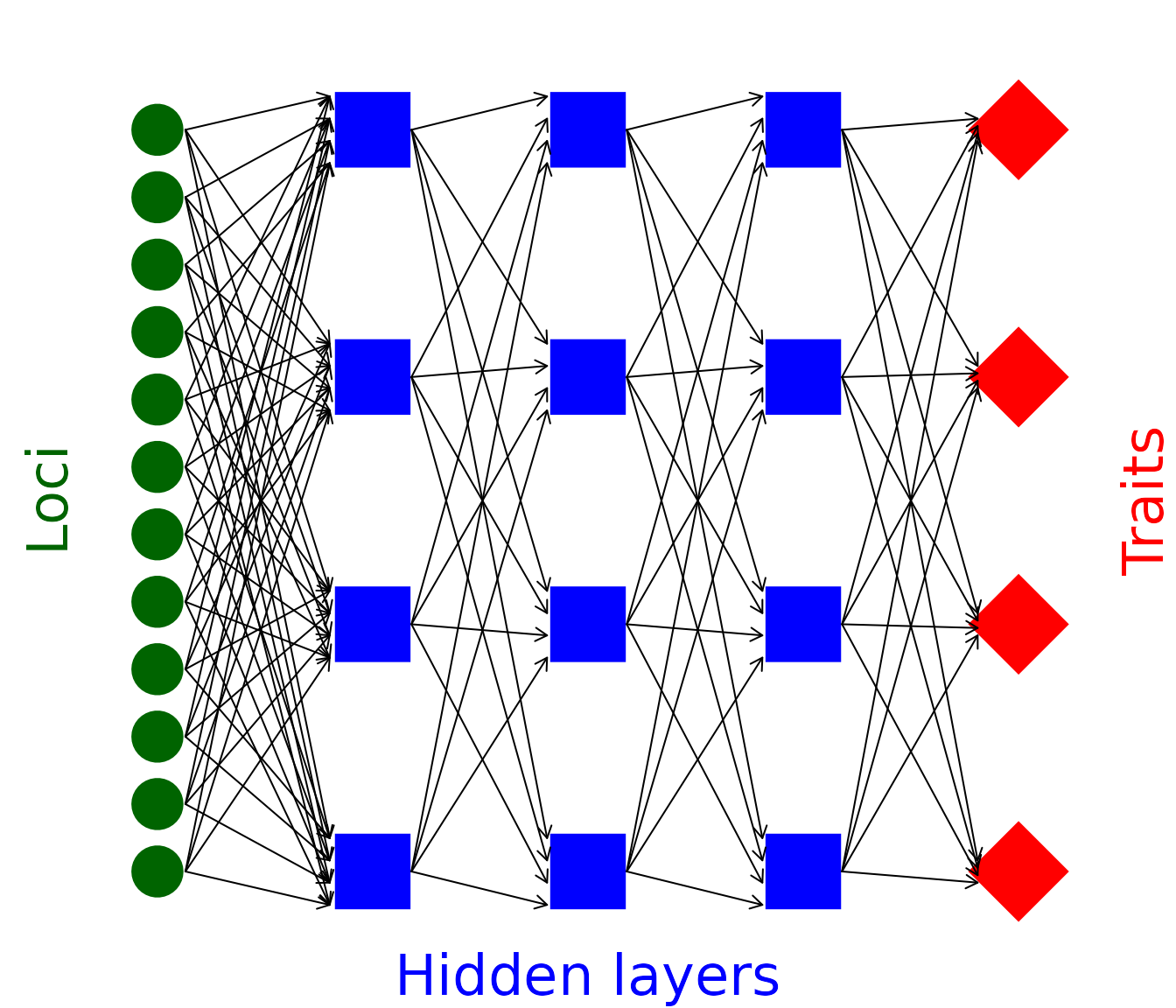
Figure 1: Network mapping loci to traits through an intermediate set of hidden layers in the mine_gmatrix function
The objective of the evolutionary algorithm in resevol is to find a
set of network values that produce traits (red diamonds above) with the
pre-specified covariance structure given allele values (green circles
above) that are sampled from a standard normal distribution (i.e., mean
of zero and standard deviation of one). Conceptually, the problem is
simple; we need to find values for the black arrows above that produce
traits that covary in the way that we want them to covary (within some
acceptable margin of error). The mine_gmatrix function uses
an evolutionary algorithm for identifying sets of values that work. An
evolutionary algorithm is an algorithm that works especially well for
I know it when I see it problems (Hamblin 2013). Luke (2013) explains
the idea behind these algorithms in more detail:
They’re algorithms used to find answers to problems when you have very little to help you: you don’t know beforehand what the optimal solution looks like, you don’t know how to go about finding it in a principled way, you have very little heuristic information to go on, and brute-force search is out of the question because the space is too large. But if you’re given a candidate solution to your problem, you can test it and assess how good it is. That is, you know a good one when you see it.
In the mine_gmatrix function of the resevol package, an
evolving population of networks like that in Figure 1 above is
initialised. Parent networks produce offspring networks with some
recombination and mutation, and offspring networks are selected based on
how close their trait covariance matrix is to the pre-specified matrix
input into the function. The algorithm is inspired by a similar
algorithm within the GMSE R package
(Duthie et al.
2018).
Key data structures used
In the code, the arrows in the above Figure 1 are represented by a set of matrices that map loci values to trait values. There are 12 loci in Figure 1, and 4 nodes in each hidden layer (blue squares). Arrow values between loci and the first hidden layer can then be represented by a matrix with 12 rows and 4 columns (i.e., row 1 holds a value for each of 4 arrows that point to the 4 hidden layer nodes). Note that the values below are initialised randomly, which is how they are initialised in the evolutionary algorithm.
arrows_1_dat <- rnorm(n = 4 * 12, mean = 0, sd = 0.1);
arrows_1_mat <- matrix(data = arrows_1_dat, nrow = 12, ncol = 4);
print(arrows_1_mat);## [,1] [,2] [,3] [,4]
## [1,] -0.1400043517 0.206502490 0.188850493 -0.191008747
## [2,] 0.0255317055 -0.163098940 -0.009744510 -0.027923724
## [3,] -0.2437263611 0.051242695 -0.093584735 -0.031344598
## [4,] -0.0005571287 -0.186301149 -0.001595031 0.106730788
## [5,] 0.0621552721 -0.052201251 -0.082678895 0.007003485
## [6,] 0.1148411606 -0.005260191 -0.151239965 -0.063912332
## [7,] -0.1821817661 0.054299634 0.093536319 -0.004996490
## [8,] -0.0247325302 -0.091407483 0.017648861 -0.025148344
## [9,] -0.0244199607 0.046815442 0.024368546 0.044479712
## [10,] -0.0282705449 0.036295126 0.162354888 0.275541758
## [11,] -0.0553699384 -0.130454355 0.011203808 0.004653138
## [12,] 0.0628982042 0.073777632 -0.013399701 0.057770907We can initialise 12 allele values, one for each locus.
loci <- rnorm(n = 12, mean = 0, sd = 1);To then get the value of the first column of four hidden layer nodes (i.e., the first column of blue squares in Figure 1), we can use matrix multiplication.
## [,1] [,2] [,3] [,4]
## [1,] -0.2918091 0.4414658 0.3604707 0.609829We can likewise use a 4 by 4 square matrix to represent the values of the arrows from the first column of four hidden layer nodes to the second column of hidden layer nodes.
arrows_2_dat <- rnorm(n = 4 * 4, mean = 0, sd = 0.1);
arrows_2_mat <- matrix(data = arrows_2_dat, nrow = 4, ncol = 4);
print(arrows_2_mat);## [,1] [,2] [,3] [,4]
## [1,] 0.10743459 -0.11775633 0.04886288 0.13373204
## [2,] -0.06650882 -0.09758506 -0.16994506 0.02366963
## [3,] 0.11139524 0.10650573 -0.14707363 0.13182934
## [4,] -0.02458964 0.01316706 0.02841503 0.05239098We can then use matrix multiplication to map the 12 allele values to the values of the second column of hidden layer nodes.
## [,1] [,2] [,3] [,4]
## [1,] -0.03555252 0.03770376 -0.124971 0.05089525This pattern can continue, with 4 by 4 square matrices representing the value of arrows between columns of hidden layer nodes, and between the last hidden layer column and traits (note that the number of hidden layer columns can be any natural number, but the number of nodes within a column always equals the number of traits). In the actual evolutionary algorithm code, all of these square matrices are themselves held in a large 3D array. But the idea is the same; a vector of allele values is multiplied by multiple matrices until a set of trait values is produced. If multiple vectors of random standard normal allele values are generated, then the traits that they produce from all of this matrix multiplication can be made to covary in some pre-specified way using the evolutionary algorithm.
General overview of the evolutionary algorithm
The evolutionary algorithm first initialises a population of networks, with each network having a unique set of values (i.e., black arrows in Figure 1, represented in the code by matrices explained in the previous selction). In each iteration of the evolutionary algorithm, with some probability, networks crossover their values with another randomly selected network. Individual values in each network then mutate with some probability. The fitness of each network is then calculated by comparing its trait covariances with those of a pre-specified covariance matrix. A tournament is then used to select the highest fitness networks, and those selected networks replace the old to comprise the new population. Iterations continue until either a maximum number of iterations is reached or a network is found that produces trait covariances sufficiently close to the pre-specified covariance matrix. Figure 2 below provides a general overview of the evolutionary algorithm.
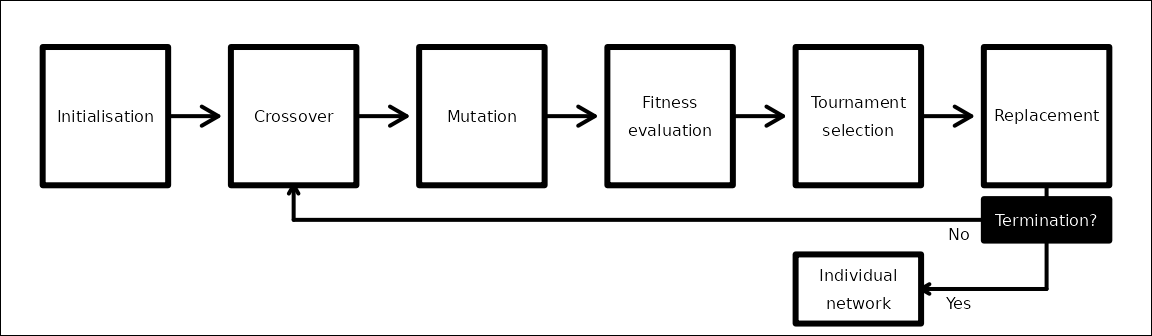
Figure 2: Conceptual overview of the evolutionary algorithm used in the resevol package.
The steps listed in the box above are explained in more detail below
with reference to the arguments applied in the mine_gmatrix
function that calls it.
Initialisation
At the start of the evolutionary algorithm, a population of
npsize networks is initialised. Each individual network in
this population is represented by one matrix and one three dimensional
array (see an explanation of the data
structures above). All the elements of networks are initialised with
a random sample from a normal distribution with a mean of 0 and a
standard deviation of sd_ini.
Crossover
An iteration of the evolutionary algorithm begins with a crossover
between the values of networks in the population. For each network in
the population, a crossover event will occur in the values linking loci
to the first hidden layer (see Figure 1) with a probability of
pr_cross. And a second independent crossover event will
occur in the values linking the first hidden layer to traits, also with
a probability of pr_cross. The reason that these two
crossover events are independent is due to the different dimensions of
the underlying arrays (see key data structures
used above). A matrix with loci rows and a number of
columns that matches the number of traits holds the values linking loci
to the first hidden layer. A three dimensional array with row and column
numbers matching trait number, and a depth matching the number of hidden
layers (e.g., 3 in Figure 1) holds the remaining values
linking the first hidden layer to trait values.
If a crossover event occurs for a focal network, then a contiguous set of values is defined and swapped with another randomly selected network in the population. Dimensions of the contiguous set are selected from a random uniform distribution. For example, given that the network in Figure 1 would be represented by a three dimensional array with 4 rows, 4 columns, and 3 layers, three random integers from 1-4, 1-4, and 1-3 would be sampled twice, respectively, with replacement. If the values selected were 1, 3, and 2 in the first sample, then 3, 3, and 1 in the second sample, then all values from rows 1-3, column 3, and layers 1-2 would be swapped between networks. Conceptually, this is the equivalent of drawing a square around set of arrows in Figure 1 and swapping the arrow values with the values of another network.
Mutation
After crossover occurs, all network values mutate independently at a
fixed probability of mu_pr. If a mutation event occurs,
then a new value is randomly sampled from a normal distribution with a
mean of 0 and a standard deviation of mu_sd. This value is
then added to the existing value in the network.
Fitness evaluation
After mutation, the fitness of each network in the population is
evaluated. For each network, a set of indivs loci vectors
is created, which represents the allele values of indivs
potential individuals in a simulated population. Elements of each loci
vector are randomly sampled from a standard normal distribution. For
example, in the network of Figure 1 where loci = 12,
loci * indivs standard normal values would be generated in
total. After these indivs loci vectors are initialised,
values in each vector are mapped to traits using the focal network,
thereby producing indivs sets of traits from the focal
network. These indivs sets of traits are then used to
calculate the among trait covariances and build a trait covariance
matrix for the network. This trait covariance matrix is then compared to
the pre-specified gmatrix by calculating stress as the mean
squared deviation between matrix elements. Lower stress values
correspond to higher network fitness.
The stress of each network in the population of npsize
networks is calculated using the above algorithm. Selection of the next
generation of npsize networks is then done using a
tournament.
Tournament selection
After fitness evaluation, networks in the population compete in a
series of tournaments to determine the composition of the next
generation of npsize networks. Tournament selection is a
flexible way to choose the fittest subset of the population (Hamblin 2013).
It starts by randomly selecting sampleK networks with
replacement to form the competitors in a tournament (note that
sampleK is constrained to be less than or equal to
npsize). Of those sampleK networks, the
chooseK networks with the highest fitness (i.e., lowest
stress) are set aside to be placed in the new population (note that
chooseK must be less than or equal to
sampleK). More tournaments continue until a total of
npsize new networks are set aside to form the new
generation of networks.
Termination
Throughout the evolutionary algorithm, the network with the lowest
overall stress (from any generation) is retained. The evolutionary
algorithm terminates if either the logged stress of the mean network is
less than or equal to term_cri, or if max_gen
generations of the evolutionary algorithm have occurred. The mean
network stress is used instead of the lowest overall stress because
error in randomly generated loci can result in unusually low stress
values due to chance, which might not be replicated with a new random
sample of loci. When the evolutionary algorithm terminates, only the
network with the lowest overall stress is returned.
Haploid and diploid individuals
The evolutionary algorithm does not distinguish between haploid and
diploid genomes Instead, haploid and diploid individuals in resevol
simulations are build differently from the mined network described
above. For haploid individuals, network values are placed in individual
genomes exactly as they are returned by mine_gmatrix.
Hence, standard normal allele values at loci from haploid genomes will
map to predictably covarying traits. For diploid individuals, all
network values returned by mine_gmatrix are divided by 2,
and two copies of each value are then placed in each individual to model
diploid genomes. Allele values are then randomly sampled from a normal
distribution with a mean of 0 and a standard deviation of
1 / sqrt(2), so that summed allele values at homologous
loci will have a standard normal distribution. As such, effects of each
loci are determined by the sum of homologous alleles. Similarly,
homologous network values mapping allele values to traits are also
summed, thereby producing the expected trait covariance structure.